GLP-1R is a class B GPCR centrally involved in glucose homeostasis, insulin secretion, appetite, and β-cell maintenance and thus stands as one of the most important targets in metabolic disease research and modern incretin-based therapeutics.1 How various peptide ligands engage this receptor is central to understanding their pharmacological behavior and underpinning drug discovery efforts focused on diabetes, obesity, and emerging metabolic disorders.2
The peptide-based drugs studied here fall into two families with distinct biological origins and structural properties. Specifically, the first family is comprised of the parent peptide GLP-1, being the endogenous incretin hormone3, whereas liraglutide and semaglutide are the clinically optimized analogs of GLP-1, which have been engineered for enhanced stability and protracted action through certain structural changes such as conjugation with a fatty-acid chain.4 The other family was made up of exendin-4 and exendin-9, from a separate evolutionary lineage, with exendin-4 serving as a strong agonist while the truncated form, exendin-9, acts as an antagonist competing for receptor binding but not triggering any further signaling.5&6
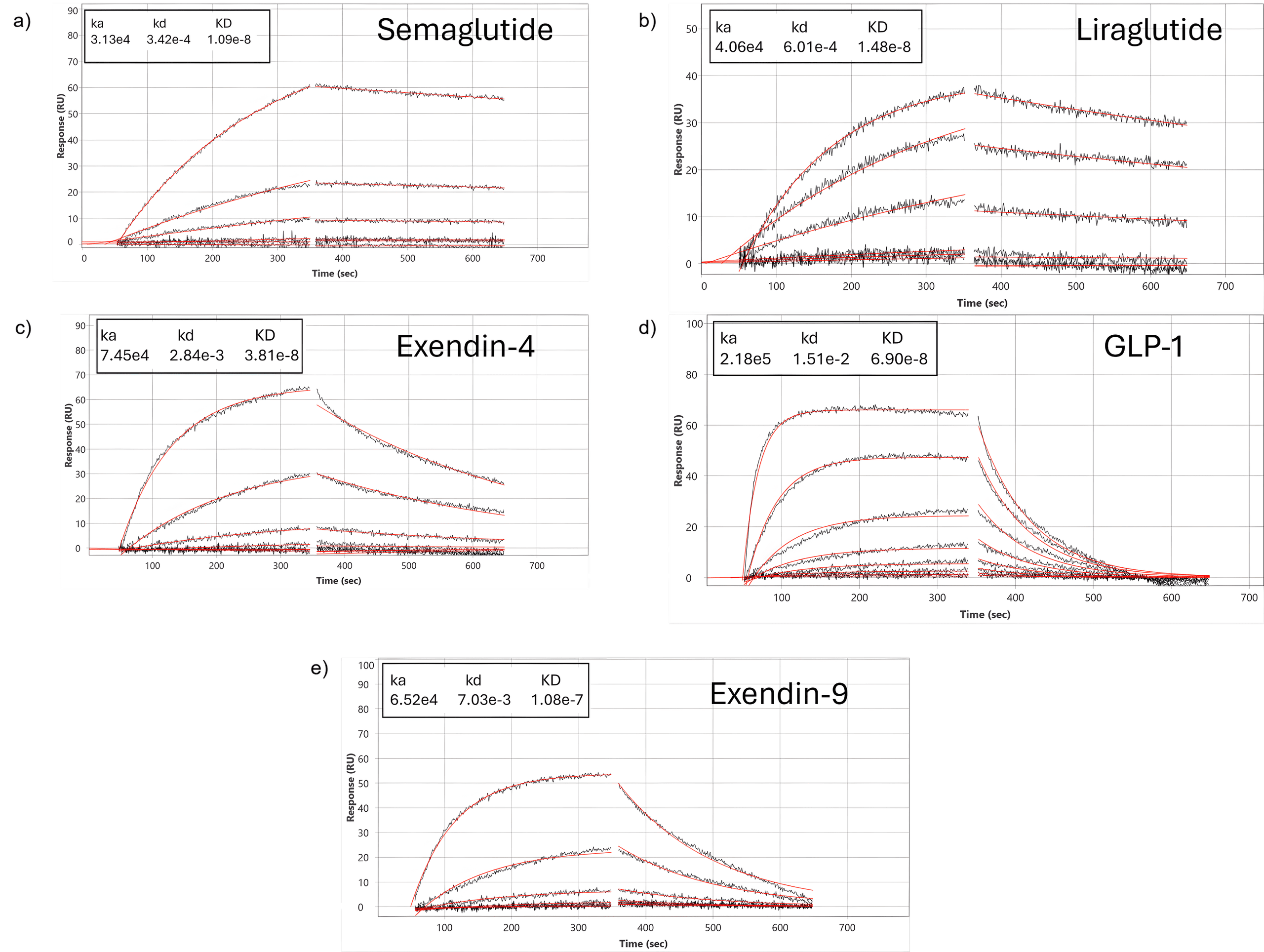
Figure 1: Sensorgrams of semaglutide, liraglutide, exendin-4, GLP-1 and exendin-9 interacting with an extracellular domain of GLP-1R surface. Each compound was injected using a 3-fold dilution series. The entire data set fits globally with a 1:1 interaction model as shown by red lines
The 5-channel BI-4500 SPR provides an ideal platform to resolve these differences because it allows for real-time, label-free quantification of kinetic parameters. By immobilizing only, the GLP-1R extracellular domain (ECD) on the sensor surface, the assay isolates the critical initial binding step common to all GLP-1R peptide ligands, independent of contributions from the receptor’s transmembrane domains or intracellular signaling machinery. The resulting configuration allows kinetic rate constants and apparent affinities to be precisely determined, revealing subtle differences in overall binding strengths that help explain the distinct pharmacological profiles of each peptide. As these measurements are made in real time under flow, SPR uniquely resolves subtle kinetic differences that are often invisible in endpoint radioligand or fluorescence assays and is therefore well suited to rank early-stage drug candidates, benchmark them against marketed drugs and serve as a guide for subsequent in cell studies.
The distinct secondary structures and sequence modifications within the GLP-1 glutide and exendin peptide families directly influence their binding affinities for the GLP-1 receptor ECD, reflected clearly in their measured KD values: semaglutide (10 nM), liraglutide (14 nM), exendin-4 (38 nM), GLP-1 (69 nM) and exendin-9 (108 nM).
| Peptides | Alpha helix % in buffer | KD (nM) |
| Semaglutide* | 43 ± 0.9 | 10 ± 3.8 |
| Liraglutide* | 42 ± 0.6 | 14 ± 2.8 |
| Exendin-4** | 40 | 38 ± 9.3 |
| GLP-1* | 34 ± 1.5 | 69 ± 10.1 |
| Exendin-9** | 15 | 108 ± 7.8 |
Table 1: Alpha helix % of peptides and kinetic values of peptide binding to GLP-1R. Peptides marked with a single asterisk (*) indicate alpha-helical values obtained from RedShiftBio microfluidic7 modulation spectroscopy measurements, whereas those marked with a double asterisk (**) indicate alpha-helical values obtained from other published literature sources.8 KD values from BI represent equilibrium dissociation constants for the human GLP-1 receptor, with lower KD indicating higher binding affinity.
Semaglutide and liraglutide show the highest alpha helical content in the buffer among the ligands studied. They also demonstrate the strongest binding to GLP-1R. Their increased helicity is expected to stabilize the peptides in shapes that are organized for high-affinity engagement with the ECD, which lowers the entropic cost of receptor binding. Native GLP-1 has a lower amount of alpha helix content and therefore would be expected to have less structural stability and weaker affinity compared to its analogues, which is indeed observed in data shown in Table 1. Additionally, exendin-4 has an alpha helical content like the GLP-1 analogues, but it has only moderate ECD binding and is weaker than the glutides which could be due to a difference in the C-terminal fold that does not fit well with the GLP-1R binding site. Exendin (9-39), the shorter antagonist version of exendin-4, is missing key N-terminal residues needed for stable helix formation and receptor activation. As a result, it shows the weakest affinity in this series. Overall, these findings show how intrinsic helical structure and ligand organization majorly shape binding interactions in the GLP-1 receptor system.
These kinetic signatures, when captured by SPR, enable quantitative comparisons among peptide classes underpinning structure-activity relationship analyses, and help improve understanding of how structural modifications to therapeutic peptides affect receptor engagement. In summary, SPR measurements of the GLP-1R ECD provide a high-resolution tool for distinguishing ligand classes and furthering the optimization of next-generation GLP-1R therapeutics.
Author: Nguyen Ly, and Miyuki Thirumurthy | Biosensing Instrument | Published February 25, 2026
DOWNLOAD PDF
Download a PDF of Application Note 174: Structure-Dependent ECD Anchoring of GLP-1R by Therapeutic Peptides
- Drucker, Daniel J et al. Cell Metabolism 27, no. 4 (2018): 740-756.
- Deganutti, et al. Nature Communications 13, no. 1 (2022): 92.
- Holst, J. J et al, Physiological Reviews (2007).
- Sangwung, et al, American Journal of Physiology-Endocrinology and Metabolism 327, no. 5 (2024): E600-E615.
- Eng et al, Journal of Biological Chemistry 267, no. 11 (1992): 7402-7405.
- Schirra et al,The Journal of Clinical Investigation 101, no. 7 (1998): 1421-1430.
- Collins,Valerie, Richard Huang, RedShiftBio Application Note 850-0147 (2025) https://www.redshiftbio.com/resources/glp-1-analogue-structures-formulations-via-mms2
- Andersen et al Bioorganic & Medicinal Chemistry 10, no. 1 (2002): 79-85.